History
Mr. Tsang was a 20-year-old gentleman. He was a non-smoker and non-drinker. He had experienced a normal perinatal process and developmental milestones. His educational level was up to higher diploma.
He had no known past medical conditions, including significant head trauma, central nervous system infections or febrile seizures.
He did not have a family history of epilepsy.
He presented with acute-onset intermittent abnormal left finger movements for 3 days. The movements were brief and may occur in clusters. Left-hand function was otherwise normal during asymptomatic intervals. There were no obvious precipitating factors.
He did not experience fever, headache, neck pain, altered consciousness. He denied the use of illicit drugs.
Physical examination
Neurological examination was pertinent for brief, jerky, non-rhythmic movements of the left-sided fingers. These movements were consistent with polyminimyoclonus.
Power of the limbs was normal. A left-sided hyperreflexia was demonstrated.
There were no features of neurocutaneous disorders.
Investigations
Baseline blood tests, syphilis serology, inflammatory markers, autoimmune markers, ceruloplasmin, ammonia, and lactate were normal.
A plain CT brain revealed asymmetrical cerebral atrophy worse over the right hemisphere, predominantly affecting the frontal / temporal / insular regions, with proportional enlarged bilateral lateral and third ventricles. There were no gyral calcifications.
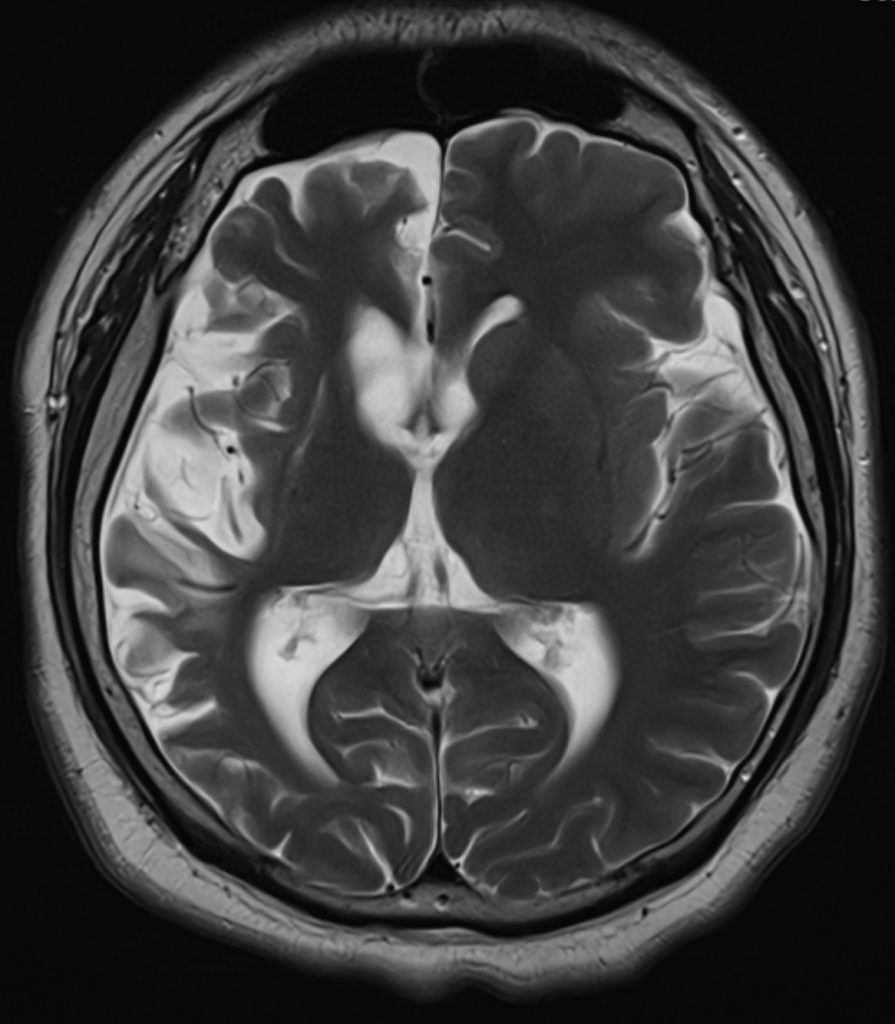
A contrast MRI brain illustrated a similar right hemisphere predominant atrophy, with mild T2W/ FLAIR hyperintense change over the atrophic right lentiform nucleus and right cerebral cortex. There were no leptomeningeal enhancement.
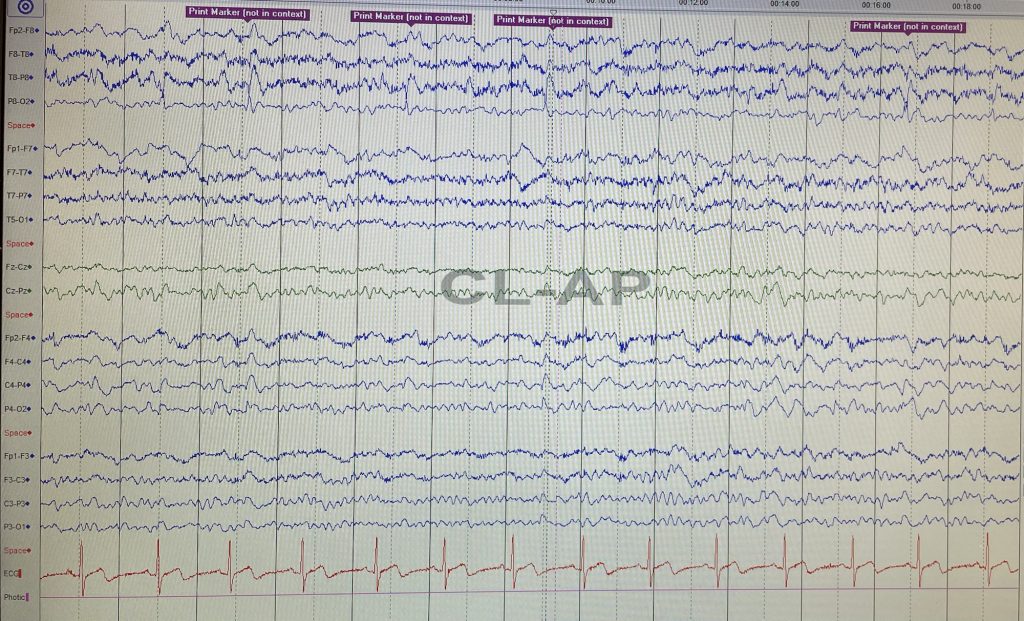
An EEG showed asymmetrical background with right hemispheric slowing. There were sharp-slow and spike-slow waves over the right posterior temporal regions with concomitant left finger polyminimyoclonus.
CSF specimens were not suggestive of meningeal inflammation.
Thought process
The pathology was likely localized to the cerebrum.
The self-remitting, episodic polyminimyoclonus were likely ictal events that had electrographic correlates.
The global, albeit asymmetrical, cerebral atrophy was a sequelae of a prior insult during brain development, the nature of which was uncertain. The insult predominantly affected the non-dominant lobe, thus might not have manifested clinically.
Therefore, this would be a case of remote symptomatic cortical myoclonic epilepsy.
There were no evident clues suggestive of a neurocutaneous syndrome. An apparently normal intellect would argue against this differential etiology.
Consideration was made to the possibility of Rasmussen encephalitis. The clinical course was deemed to be atypical. Given the radiological findings, the patient should be expected to have intractable epilepsy and severe focal neurological deficits as in the active stage of the disease, which was not the case.
Next steps
Levetiracetam was initiated. An MRI brain was arrange to monitor for serial progression of the asymmetrical cerebral atrophy.